Overview
TopRepo is a comprehensive top-down proteomics (TDP) spectral
repository generated from 3,165 MS raw files across 33 publications.
The repository comprises 12,879,563 tandem mass spectrometry (MS/MS)
spectra from 12 species, 7 types of mass spectrometers and 4
dissociation methods, and 5,147,250 proteoform-spectrum matches
(PrSMs).
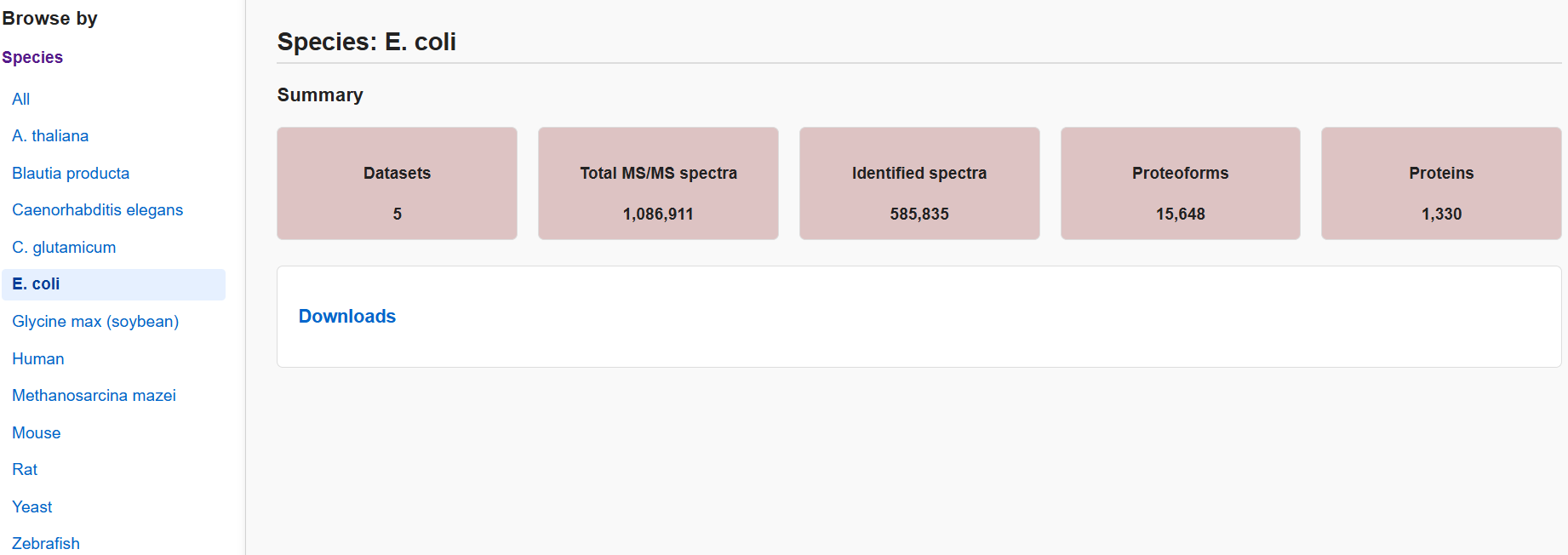
All datasets are organized by species in TopRepo.
Please go to Download page to browse
and download datasets.
File format
The datasets in TopRepo includes multiple file formats to support diverse applications.
- Raw files contain the original mass spectrometry data, which are stored in their respective resposities (PRIDE or MASSIVE).
- Metadata TSV files provide experimental details and acquisition parameters for each MS data file.
- Spectral TSV files contain spectral identifications reported by TopPIC in tab-separated format.
- MSALIGN files provide deconvoluted MS/MS spectra with fragment mass annotations.
- MGF files contain centroided spectra and annotatations of deconvoluted masses and matched fragment ions.
1. Raw file
Raw files contain MS data produced by mass spectrometers. Due to storage limitation, the raw files of TopRepo are not hosted directly on this website. Instead, the raw files are provided through their original public repositories, either PRIDE or MASSIVE. The links to all associated raw files of TopRepo are provided in the table.
2. Metadata TSV file
The metadata file includes MS experimental information for each raw data file in the TSV format, including sample types, mass spectrometers, separation methods, and fragmentation techniques. It also provides statistics of the data file, such as the numbers of spectra, PrSM, proteoform, and protein identifications.
| Column | Description |
|---|---|
| Dataset_id | Unique identifier of the dataset (PRIDE or MASSIVE). |
| Raw_file_name | Name of raw mass spectrometry data file. |
| MZML_file_name | Name of the converted mzML file by msconvert. |
| #Msalign_files | Number of MSALIGN files generated by TopFD. |
| #MS/MS_spectra | Total number of MS/MS spectra in this file. |
| #PrSM_ids | Number of peptide-spectrum matches (PrSM) identified by TopPIC. |
| #Proteoform_ids | Number of proteoform identified in this file. |
| #Protein_ids | Number of proteins identified in this file. |
| Species | Species of the sample. |
| Instrument | MS instrument used for data acquisition. |
| Dissociation | Fragmentation method used for MS/MS (e.g., HCD, CID, ETD, EtHCD). |
| Sample | Description of the biological sample. |
| Separation | Separation techinque applied prior to MS analysis (e.g., CZE, RPLC). |
| Cystein_protection | Chemical treatment applied to cystein residues (e.g. carbamidomethylation, none). |
| FAIMS | Field Asymmetric Ion Mobility Spectrometry is used or not. |
3. Spectral TSV file
This file contains detailed spectrum-level identification results, integrating information from mzML, MSALIGN files, and TopPIC output TSV files.
| Column | Description |
|---|---|
| PROJECT_id | Unique identifier of the project. |
| SUBDATASET_id | Unique identifier of the subdataset. |
| DATASET_id | Unique identifier of the project to which this spectrum belongs. |
| MZML_file_name | The mzML MS file name. |
| MZML_instrument | Instrument used to acquire the MS data. |
| MZML_ms1_scan | Scan number of the MS1 spectrum in the mzML file. |
| MZML_ms1_scan_window_lower_limit | Lower m/z boundary of the MS1 scan window. |
| MZML_ms1_scan_window_upper_limit | Upper m/z boundary of the MS1 scan window. |
| MZML_ms1_retention_time | Retention time of the MS1 scan. |
| MZML_ms1_total_ion_current | Total ion current of the MS1 spectrum. |
| MZML_ms1_mass_resolving_power | Mass resolution of the MS1 scan. |
| MZML_ms1_ion_injection_time | Ion injection time used for the MS1 scan. |
| MZML_ms1_lowest_observed_mz | Lowest observed m/z value in the MS1 spectrum. |
| MZML_ms1_highest_observed_mz | Highest observed m/z value in the MS1 spectrum. |
| MZML_ms2_scan | Scan number of the MS2 spectrum. |
| MZML_ms2_scan_window_lower_limit | Lower m/z boundary of the MS2 scan window. |
| MZML_ms2_scan_window_upper_limit | Upper m/z boundary of the MS2 scan window. |
| MZML_ms2_retention_time | Retention time of the MS2 scan. |
| MZML_ms2_total_ion_current | Total ion current of the MS2 spectrum. |
| MZML_ms2_mass_resolving_power | Mass resolution of the MS2 scan. |
| MZML_ms2_ion_injection_time | Ion injection time used for the MS2 scan. |
| MZML_ms2_lowest_observed_mz | Lowest observed m/z value in the MS2 spectrum. |
| MZML_ms2_highest_observed_mz | Highest observed m/z value in the MS2 spectrum. |
| MZML_isolation_window_target_mz | Target m/z value of the isolation window for precursor selection. |
| MZML_isolation_window_lower_offset | Lower offset of the isolation window relative to the target m/z. |
| MZML_isolation_window_upper_offset | Upper offset of the isolation window relative to the target m/z. |
| MZML_selected_ion_mz | M/Z value of the selected precursor ion. |
| MZML_selected_ion_peak_intensity | Intensity of the selected precursor ion. |
| MZML_selected_ion_charge | Charge state of the selected precursor ion. |
| MZML_activation | Fragmentation method used for MS/MS (e.g., HCD, CID, ETD). |
| MZML_collision_energy | Collision energy used in fragmentation. |
| MSALIGN_file_name | Name of the MSALIGN file. |
| MSALIGN_ms1_id | ID of the MS1 spectrum in the MSALIGN file. |
| MSALIGN_ms2_id | ID of the MS2 spectrum in the MSALIGN file. |
| MSALIGN_precursor_charge | Charge states of the precursor ions reported in MSALIGN. |
| MSALIGN_precursor_monoisotopic_mass | Monoisotopic masses of the precursor ions. |
| MSALIGN_precursor_intensity | Intensities of the precursor ions. |
| MSALIGN_precursor_feature_id | Feature ID associated with the precursor ion. |
| MSALIGN_precursor_feature_intensity | Intensity of the precursor feature. |
| MSALIGN_precursor_feature_score | Score assigned to the precursor feature. |
| MSALIGN_precursor_feature_apex_time | Retention time at the apex of the precursor feature. |
| MSALIGN_number_of_fragment_ions | Number of fragment ions in the MS2 spectrum. |
| TOPPIC_prsm_id | Unique ID of the PrSM reported by TopPIC. |
| TOPPIC_adjusted_precursor_mass | Adjusted precursor mass after TopPIC calibration. |
| TOPPIC_proteoform_id | ID of the identified proteoform. |
| TOPPIC_proteoform_intensity | Total signal intensity across multiple scans and charge states associated with the proteoform. |
| TOPPIC_number_of_protein_hits | Number of proteins containing the proteoform sequence matched to this spectrum. |
| TOPPIC_protein_accession | Accession number of the matched protein. |
| TOPPIC_protein_description | Description of the matched protein. |
| TOPPIC_first_residue_position | Start position (1-based) of the proteoform within the protein sequence. |
| TOPPIC_last_residue_position | End position (1-based) of the proteoform within the protein sequence. |
| TOPPIC_special_amino_acids | List of special amino acids present in the proteoform. |
| TOPPIC_database_sequence | Amino acid sequence of the proteoform identification without annotations of modifications. |
| TOPPIC_proteoform_mass | The monoisotopic molecular mass of the proteoform. |
| TOPPIC_protein_n-terminal_form | N-terminal form of the protein (e.g., NME and NTA). |
| TOPPIC_fixed_modifications | List of fixed post-translational modifications. |
| TOPPIC_number_of_unexpected_modifications | Number of unexpected modifications detected. |
| TOPPIC_unexpected_modifications | Description of unexpected modifications. |
| TOPPIC_number_of_variable_modifications | Number of variable post-translational modifications. |
| TOPPIC_variable_modifications | Description of variable post-translational modifications. |
| TOPPIC_miscore | MIScore for modification localization reported by TopPIC. |
| TOPPIC_number_of_matched_experimental_fragment_ions | Number of matched experimental fragment ions in the MSALIGN file. |
| TOPPIC_number_of_matched_theoretical_fragment_masses | Number of matched theoretical fragment masses of the proteoform. |
| TOPPIC_e-value | E-value of the PrSM. |
| TOPPIC_spectrum_level_q-value | Q-value at the spectrum level. |
| TOPPIC_proteoform_level_q-value | Q-value at the proteoform level. |
| TOPPIC_proteoform | Annotated proteoform sequence. |
| TOPPIC_previous_residue | Amino acid residue preceding the proteoform in the protein sequence. |
| TOPPIC_next_residue | Amino acid residue following the proteoform in the protein sequence. |
4. MSALIGN file
The MSALIGN file includes spectral information, its identified proteoform, deconvoluted fragment ions, and their annotations for each MS/MS spectrum.
| Line | Description |
|---|---|
| BEGIN IONS | Start of an MS/MS spectrum block. Marks the beginning of a new spectrum entry. |
| DATASET_ID | Unique identifier of the dataset. |
| MZML_FILE_NAME | Name of the mzML file containing the MS data. |
| MSALIGN_FILE_NAME | Name of the MSALIGN file. |
| MS2_ID | Index of the MS2 spectrum within the file. |
| MS2_SCAN | Scan number of the MS2 spectrum. |
| MS2_RETENTION_TIME | Retention time (in seconds) of the MS2 scan. |
| LEVEL | MS level of the spectrum (level=2 for all entries). |
| MS1_ID | Index of the associated MS1 spectrum. |
| MS1_SCAN | Scan number of the associated MS1 spectrum. |
| PRECURSOR_WINDOW_BEGIN | Lower bound of the precursor isolation window (m/z). |
| PRECURSOR_WINDOW_END | Upper bound of the precursor isolation window (m/z). |
| ACTIVATION | Fragmentation method used (e.g., HCD, CID, ETD). |
| PRECURSOR_MONOISOTOPIC_MZ | Monoisotopic m/z values of the precursor ions. The isolation window may contain multiple precursor ions. Multiple m/z values are separated by the ":" symbol. |
| PRECURSOR_CHARGE | Charge states of the precursor ions. |
| PRECURSOR_MONOISOTOPIC_MASS | Monoisotopic masses of the precursor ions. |
| PRECURSOR_INTENSITY | Intensities of the precursor ions. |
| PRECURSOR_FEATURE_ID | IDs of the features of the precursor ions. |
| INSTRUMENT | MS instrument used for data acquisition. |
| COLLISION_ENERGY | Collision energy for MS/MS fragmentation. |
| PROTEIN_ACCESSION | Protein accession ID from the UniProt database. |
| DATABASE_SEQUENCE | Amino acid sequence of the matched protein from the database. |
| FIRST_RESIDUE_POSITION | Position (1-based) of the first amino acid residue of the identified proteoform in the protein sequence. |
| PROTEOFORM | Proteoform sequence with annotations of modifications. |
| FIXED_MODIFICATIONS | List of fixed PTMs, including positions and mass shifts. |
| UNEXPECTED_MODIFICATIONS | List of unexpected PTMs, including position and mass shifts. |
| E_VALUE | Estimated E-value of the identification. |
| SEQUENCE_COVERAGE | The number of peptide bonds covered by matched fragment ions of the identified proteoform. |
| END IONS | End of an MS/MS spectrum block. |
Each deconvoluted fragment ion is represented by 10 columns in a line.
| Column | Description |
|---|---|
| 1. mass | Fragment ion monoisotopic mass. |
| 2. intensity | Fragment ion intensity. |
| 3. charge | Charge state of the fragment ion. |
| 4. confidence | Confidence score of the deconvoluted fragment ion. |
| 5. ion-type | Type of matched theoretical fragment ion (e.g., b or y). |
| 6. number of residues | Number of residues in the matched theoretical fragment ion. |
| 7. Position of peptide bond | Position (1-based) of peptide bond of the matched fragment ion. |
| 8. shift | Mass shift between the experimental and theoretical fragment masses. 0: no shift, +1:+1.00235 Da, -1: -1.00235 Da. |
| 9. error (Da) | Mass error in Daltons. |
| 10. error (ppm) | Mass error in parts per million (ppm). |
5. MGF file
The MGF file contains centroided peaks and additional spectral information and annotations.
| Line | Description |
|---|---|
| BEGIN IONS | Marks the beginning of a new spectrum. |
| DATASET_ID | Dataset identifier (PRIDE or MASSIVE). |
| MZML_FILE_NAME | mzML file name. |
| SCAN | Scan number of the MS/MS spectrum. |
| TITLE | Spectrum title. |
| RTINSECONDS | Retention time (in seconds) of MS/MS spectrum. |
| PEPMASS_MZ | The m/z of the selected precursor ion obtained from the mzML file. |
| CHARGE | Precursor charge state obtained from the mzML file. |
| MSALIGN_FILE_NAME | Name of the msalign file. |
| MS2_ID | Index of the MS2 spectrum within the msalign file. |
| LEVEL | MS level of the spectrum (level=2 for all entries). |
| MS1_ID | Index of the associated MS1 spectrum in the msalign file. |
| MS1_SCAN | Scan number of the associated MS1 spectrum. |
| PRECURSOR_WINDOW_BEGIN | Lower bound of the precursor isolation window (m/z). |
| PRECURSOR_WINDOW_END | Upper bound of the precursor isolation window (m/z). |
| ACTIVATION | Fragmentation method used (e.g., HCD, CID, ETD). |
| PRECURSOR_MONOISOTOPIC_MZ | Monoisotopic m/z values of the precursor ions reported in the msalign file. The isolation window may contain multiple precursor ions. Multiple m/z values are separated by the ":" symbol. |
| PRECURSOR_CHARGE | Charge states of the precursor ions reported in the msalign file. |
| PRECURSOR_MONOISOTOPIC_MASS | Monoisotopic masses of the precursor ions reported in the msalign file. |
| PRECURSOR_INTENSITY | Intensities of the precursor ions reported in the msalign file. |
| PRECURSOR_FEATURE_ID | IDs of the features of the precursor ions reported in the msalign file. |
| INSTRUMENT | MS instrument used for data acquisition. |
| COLLISION_ENERGY | Collision energy for MS/MS fragmentation. |
| PROTEIN_ACCESSION | Protein accession ID from the UniProt database. |
| DATABASE_SEQUENCE | Amino acid sequence of the matched protein from the database. |
| FIRST_RESIDUE_POSITION | Position (1-based) of the first amino acid residue of the identified proteoform in the protein sequence. |
| PROTEOFORM | Proteoform sequence with annotations of modifications. |
| FIXED_MODIFICATIONS | List of fixed PTMs, including positions and mass shifts. |
| UNEXPECTED_MODIFICATIONS | List of unexpected PTMs, including position and mass shifts. |
| E_VALUE | Estimated E-value of the identification. |
| SEQUENCE_COVERAGE | The number of peptide bonds covered by matched fragment ions of the identified proteoform. |
| END IONS | End of an MS/MS spectrum block. |
Each centroide peak is represented by 16 columns in a line.
| Column | Description |
|---|---|
| 1. m/z | Mass-to-charge ratio of the centroid peak. |
| 2. intensity | Intensity of the centroid peak. |
| 3. deconvoluted fragment ion index | Index (1-based) of the matched deconvoluted fragment ion in the msalign file. |
| 4. theoretical mass | Neutral mass of the theoretical isotopic peak of the matched deconvoluted fragment ion. |
| 5. charge | Charge state of the matched deconvoluted fragment ion. |
| 6. theoretical m/z | The m/z value of the theoretical isotopic peak of the matched deconvoluted fragment ion. |
| 7. maximum intensity in envelope (yes or no) | Indicates whether the peak is the most intense one within the theoretical isotopic envelope of the matched deconvoluted fragment ion |
| 8. index of the peak in isotopic envelope | Index (1-based) of the peak in the theoretical isotopic envelope of the matched deconvoluted fragment ion. | 9. theoretical peak intensity | Intensity of the theoretical isotopic peak of the matched deconvoluted fragment ion. |
| 10. percentage of theoretical peak intensity | Percentage of the intensity of the matched theoretical isopotic peak with respect to the total peak intensity of the isopotic envelope. |
| 11. ion-type | Type of the matched fragment ion (e.g., b or y). |
| 12. number of residues | Number of amino acid residues in the matched fragment ion. |
| 13. Position of peptide bond | Position (1-based) of peptide bond of the matched fragment ion. |
| 14. shift | Isotopic mass shift between the deconvoluted fragment ion and its matched theoretical fragment mass. 0: no shift, +1: +1.00235 Da, -1: -1.00235 Da. |
| 15. error (Da) | Mass error of the deconvoluted fragment ion in Daltons. |
| 16. error (ppm) | Mass error of the deconvoluted fragment ion in parts per million (ppm). |